DFT/TDDFT computational study of the structural,electronic and optical properties of rhodium (III) and iridium (III) complexes based on tris-picolinate bidentate ligands
1.Department of Chemistry,Dr Moulay Tahar University of Saida,Saida,Algeria
Abstract:
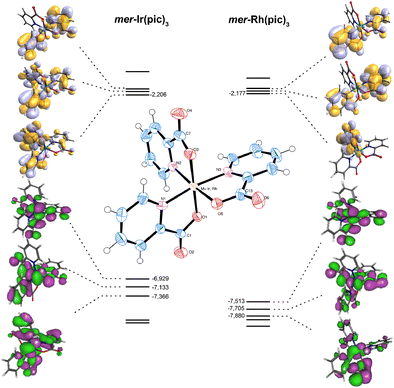
The electronic structures and spectroscopic properties of two complexes M(pic)3] (M = Ir, Rh) containing picolinate as bidentate ligands have been calculated by means density functional theory (DFT) and time-dependent DFT/TD-DFT using three hybrid functionals B3LYP, PBE0 and mPW1PW91. The PBE0 and mPW1PW91 functionals, which have the same HF exchange fraction (25%), give similar results and do not differ drastically from B3LYP results. Calculated geometric parameters of the complexes are in good agreement with the available experimental data. The UV absorptions observed in acetonitrile were assigned on the basis of singlet state transitions. The most intense band observed in the UV-C region corresponds to ligand-to-ligand charge transfer states (LLCT) in both complexes. The theoretical spectrum of the rhodium complex is characterized by a large degree of mixing between metal-to-ligand-charge-transfer (MLCT), LLCT and metal centered (MC) states in the UV-A region. The presence of low-lying excited states with MC character affects the absorption spectrum under spin-orbit coupling (SOC) effects and play important roles in the photochemical properties.