|
|||||
|
|
Naratriptan aggregation in lipid bilayers: perspectives from molecular dynamics simulations |
|
| Authors: | Irene Wood Mónica Pickholz |
| Affiliation: | 1.Instituto de Nanobiotecnología (NANOBIOTEC),Universidad de Buenos Aires, CONICET,Buenos Aires,Argentina;2.National Science Research Council (CONICET),Buenos Aires,Argentina |
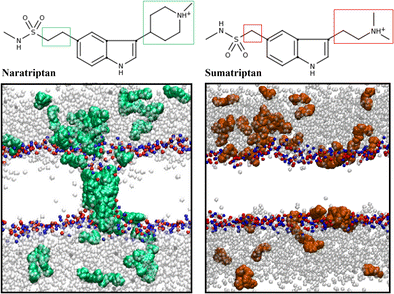
| Abstract: | In order to understand the interaction between naratriptan and a fully hydrated bilayer of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidyl-choline (POPC), we carried out molecular dynamics simulations. The simulations were performed considering neutral and protonated ionization states, starting from different initial conditions. At physiological pH, the protonated state of naratriptan is predominant. It is expected that neutral compounds could have larger membrane partition than charged compounds. However, for the specific case of triptans, it is difficult to study neutral species in membranes experimentally, making computer simulations an interesting tool. When the naratriptan molecules were originally placed in water, they partitioned between the bilayer/water interface and water phase, as has been described for similar compounds. From this condition, the drugs displayed low access to the hydrophobic environment, with no significant effects on bilayer organization. The molecules anchored in the interface, due mainly to the barrier function of the polar and oriented lipid heads. On the other hand, when placed inside the bilayer, both neutral and protonated naratriptan showed self-aggregation in the lipid tail environment. In particular, the protonated species exhibited a pore-like structure, dragging water through this environment. Graphical Abstract Different behaviour of Naratriptan and Sumatriptan, when the drugs were originally placed in the lipid core |
| Keywords: | |
| 本文献已被 SpringerLink 等数据库收录! | |