|
|||||
|
|
| 上海市崇明区猪—土壤界面微生态与抗生素耐药性相关性分析 | |
| 引用本文: | 钱璟, 吴哲元, 朱泳璋, 等. 上海市崇明区猪—土壤界面微生态与抗生素耐药性相关性分析[J]. 中国微生态学杂志, 2024, 36(2): 135-146. doi: 10.13381/j.cnki.cjm.202402002 |
| 作者姓名: | 钱璟 吴哲元 朱泳璋 张彦 郭晓奎 刘畅 |
| 作者单位: | 1. 上海交通大学医学院—国家热带病研究中心全球健康学院,上海 200025; 2. 上海交通大学医学院免疫学与微生物学系 |
| 基金项目: | 国家自然科学基金(32170141) |
| 摘 要: | 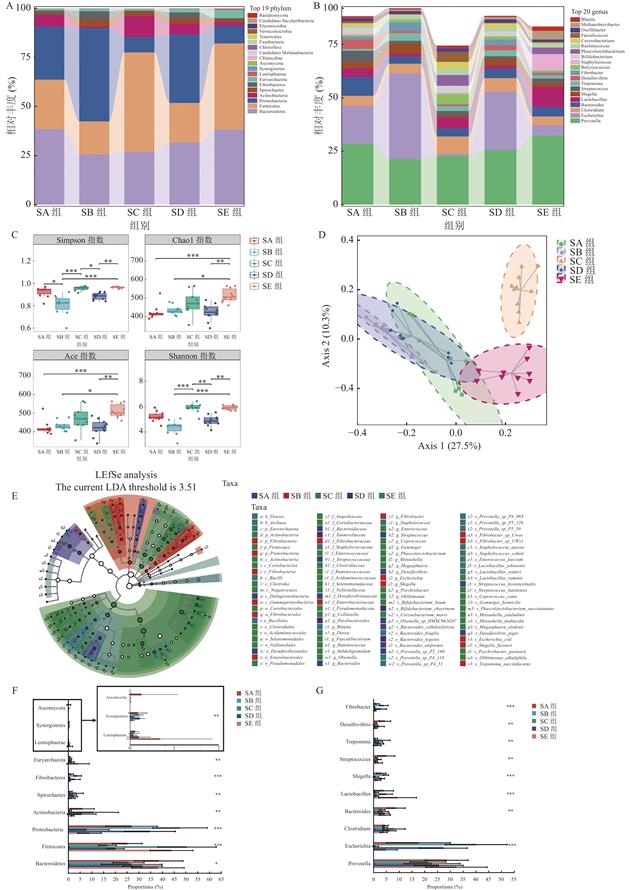 探究养殖场内育肥猪粪便与土壤的微生物组成和抗生素耐药基因构成,并挖掘之间的关联。 选择上海市崇明区5家猪养殖场,采集猪粪便和土壤样本,使用Illumina NovaSeq 6000高通量测序平台进行全基因组鸟枪法测序。通过主坐标分析、LEfSe分析、共现网络分析和统计学检验,探究样本微生物组成、多样性特征和抗生素耐药基因之间的关系。 猪粪便中的主要微生物包括拟杆菌门、厚壁菌门、变形菌门和放线菌门,主要耐药基因类型包括多药耐药类、四环素类、糖肽类、肽类、氟喹诺酮类和β-内酰胺类等。来自同一养猪场的样本在微生物丰度和耐药基因多样性上存在相关性。 通过宏基因组学方法研究了猪粪便与土壤中的微生物和耐药基因,揭示了微生物与抗生素耐药基因之间的相关性,提出微生物群是耐药基因谱的重要驱动因素。这一发现为通过微生态调控来预防和控制抗生素耐药性提供了科学依据。 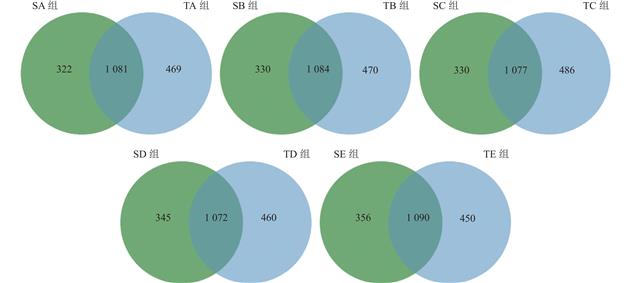 |
| 关 键 词: | 肠道微生物群 土壤微生态 微生物多样性 抗生素耐药性 宏基因组学 |
| 收稿时间: | 2023-11-02 |
| 修稿时间: | 2024-01-29 |
Correlation between microecology and antibiotic resistance at the pig-soil interface in Chongming District,Shanghai, China |
|
| QIAN Jing, WU Zheyuan, ZHU Yongzhang, et al. Correlation between microecology and antibiotic resistance at the pig-soil interface in Chongming District, Shanghai, China[J]. Chinese Journal of Microecology, 2024, 36(2): 135-146. doi: 10.13381/j.cnki.cjm.202402002 | |
| Authors: | QIAN Jing WU Zheyuan ZHU Yongzhang ZHANG Yan GUO Xiaokui LIU Chang |
| Affiliation: | 1. School of Global Health, Chinese Center for Tropical Diseases Research, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China |
| Abstract: | ObjectiveTo observe microbial composition and antibiotic resistance genes (ARGs) in pig gut and soil flora and explore the correlation between them. MethodsThis research was conducted in five swine farms in Chongming District, Shanghai. Samples of swine feces and soil were collected and subjected to Whole Genome Shotgun sequencing on the Illumina NovaSeq 6000 high-throughput sequencing platform. Through bioinformatics analysis and statistical testing, the study examined the association between microbial composition and ARGs. Results Metagenomic analysis revealed that the dominant phyla in swine manure in Chongming were Bacteroidetes, Firmicutes, Proteobacteria and Actinobacteria. Prevalent types of ARGs included multidrug, tetracycline, glycopeptide, peptide, fluoroquinolone and beta-lactam, etc. Samples from the same pig farm exhibited correlations in terms of microbial abundance and ARGs diversity. 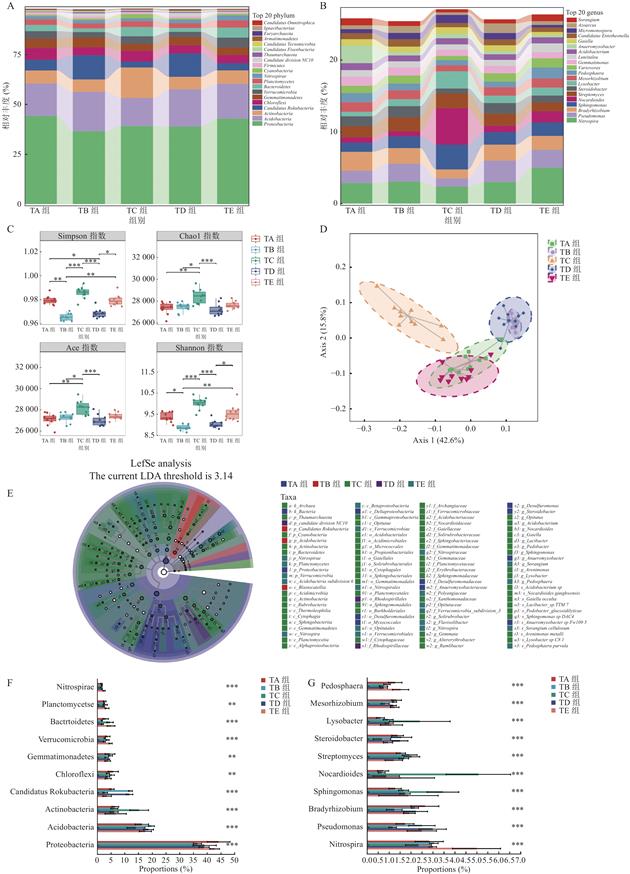 Operational factors within farms, such as feed composition, stocking density, and hygiene conditions, lead to differences in microbial composition and ARGs. At the pig feces-soil interface, beneficial bacterial taxa exhibited symbiotic relationships. These bacteria could suppress the growth of pathogenic bacteria and also had an impact on soil microecology. Various types of ARGs were found to have symbiotic relationships among themselves. ConclusionThis study investigates the composition and differences in microecology and ARGs in pig feces and soil. It reveals the close association between microbiota and ARGs, proposing that the microbiota is a significant driver of ARG profiles. This finding provides a strong basis for antibiotic resistance prevention and control through microecological regulation. |
| Keywords: | Gut microbiota Soil microbiota Microbial diversity Antibiotic resistance Metagenomics |
| 点击此处可从《中国微生态学杂志》浏览原始摘要信息 | |
| 点击此处可从《中国微生态学杂志》下载全文 | |